Ortzi Olasolo

Ortzi Olasolo
Clinical trial software must be classified correctly between GxP validation for trial support tools and SaMD regulation for software influencing patient decisions, with intended use being the key factor rather than technical complexity, since wrong classification can create major regulatory delays years later
Software has been quietly accumulating inside clinical trials for years. Some of it sits comfortably in well-understood regulatory territory — Electronic Data Capture platforms, randomization systems, electronic Trial Master Files, central lab pipelines — and is correctly handled as a GxP computerized system under ICH E6(R3), EU Annex 11, and 21 CFR Part 11. Other software does not sit so comfortably, and the volume of that other software is increasing: imaging analysis pipelines, dosimetry chains, AI-driven endpoint adjudication, decision-support tools at the investigator interface, biomarker analytics, software that determines eligibility or dose adjustment. For each of these, the sponsor and the CRO have to take a position on whether the software is a medical device or a computerized system supporting a clinical trial. The position is often taken implicitly or by default, and by the time it gets to me it has often snowballed into a serious problem.
The two regulatory frameworks lead to fundamentally different files. A GxP validation under GAMP 5 demonstrates fitness for purpose for the trial: requirements, design, IQ/OQ/PQ, audit trails, supplier qualification, change control, and sponsor oversight under ICH E6(R3). A medical device technical file demonstrates fitness for safe use in patients: ISO 14971 risk management addressing patient harm, IEC 62304 lifecycle evidence calibrated to a software safety classification, clinical evaluation under MDR Annex XIV, usability engineering under IEC 62366, post-market surveillance, and an ISO 13485 quality system behind the manufacturer. The two files overlap on traceability and configuration management, and that's it. A sponsor that has the wrong file when the question gets asked late cannot retrofit its way out of the gap on a program timeline. To further complicate things, software used in clinical trials is often embedded in a pipeline containing a mix of CE-marked or 510(k)-cleared SaMD and custom-developed software for clinical trial support. The problem here is that I often see clients trying to extend the "regulatory shield" of the CE-marked or 510(k)-cleared SaMD to the other pieces of software without much hesitation.
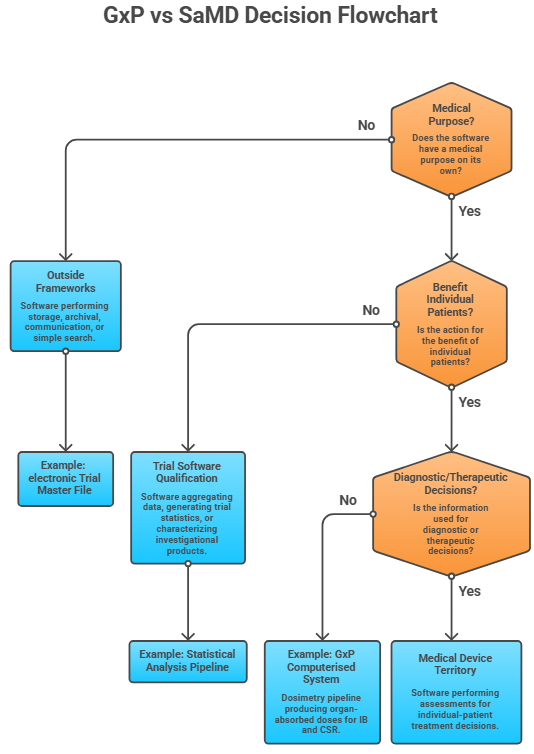
MDCG 2019-11 in the EU and the FDA framework around §201(h) and §520(o) of the FD&C Act ask broadly the same set of questions, and they all turn on intended purpose. The sequence matters because it forces the qualifier to think about who uses the output and what they do with it, rather than how sophisticated the algorithm is. The first question is whether the software has a medical purpose on its own. Software that performs storage, archival, communication, or simple search is outside the frameworks regardless of how clinically important the data being moved is. An electronic Trial Master File moving serious adverse event reports between sponsor and authority is not a medical device, even though these reports are clinically critical, because the software is not creating clinical information — it is transmitting it. A pipeline that takes raw imaging data and produces quantitative output a clinician will rely on is creating clinical information, and the medical-purpose question is answered yes.
The second question is whether the action is for the benefit of individual patients. This is where most trial software gets its qualification answer. Software that aggregates data across a population, generates trial-level statistics, or characterizes an investigational product is not acting for the benefit of individual patients even when it processes per-patient inputs. Software that returns a result to be used in the management of a specific named participant is. The distinction is not always obvious. A pharmacokinetic modeling pipeline that generates exposure- response curves for the study report is sponsor-level characterization. The same logic, embedded in a tool that gives an investigator a recommended dose adjustment for the patient sitting in front of them, has crossed the line — even though the underlying mathematics is identical.
The third question is whether the information provided is used to take decisions with diagnostic or therapeutic purposes. This is the question that catches the cases where software is genuinely informing patient management but the sponsor's narrative tries to characterize it otherwise. A central imaging review that returns tumor progression assessments to the investigator site for treatment decisions is providing information used in therapeutic decisions. The fact that it sits inside a clinical trial does not change that. Conversely, a pipeline that flags potential adverse events for safety monitoring committee review, where the safety monitoring committee takes the clinical decision based on a full data picture, is providing information that supports a sponsor-level oversight decision rather than a per-patient therapeutic one.
It is important to note that the same algorithm can have different qualification answers depending on how it is deployed, and following the information flow is usually a good indicator. Outputs flowing to the sponsor's regulatory file are not the same as outputs flowing to the investigator's screen, even when the underlying computation is identical. This also means that the qualification answer is not a property of the software in the abstract — it is a property of the intended purpose as actually documented and used in the specific trial. A tool that is correctly handled as a GxP system in one program could require a different answer in the next program if the workflow changes.
Once the test is run honestly, most software in clinical trials falls into one of three categories.
The first is software that supports trial operations. Electronic Data Capture, Randomization and Trial Supply Management, electronic Trial Master Files, statistical analysis pipelines, and electronic Clinical Outcome Assessment platforms all belong to this group. This software does not provide information about individual patients in a clinically actionable form; it manages the conduct of the trial. GxP validation, with the sponsor oversight controls in ICH E6(R3) and the data integrity controls in Annex 11, is the right and sufficient regulatory home.
The second is software that analyzes patient-level data but for sponsor-level purposes. Pharmacology characterization pipelines, exposure-response modeling, biomarker aggregation for safety review committees, dosimetry or imaging analyses that feed the Investigator's Brochure and Clinical Study Report rather than investigator-facing decisions. This software does process per-patient inputs, but its intended purpose is characterization of the investigational product, not patient management. The qualification position is defensible, but it is not automatic — it depends on a documented intended purpose, an output flow to the sponsor's regulatory file rather than to investigators, and the absence of protocol language that conditions individual patient treatment on the software's output. GxP validation is the right framework here, but the sponsor has to be willing to defend the position in detail and the documentation has to be consistent across every artifact that touches the workflow.
The third is software that provides information to investigators or treating clinicians to inform decisions about individual trial participants. Diagnostic imaging analysis returned to the site, AI- driven decision-support at the investigator interface, software that determines per-patient eligibility or dose adjustment based on its own output, real-time monitoring tools that alert investigators to clinical events. This is medical device territory under MDR Rule 11 and the FDA device definition. GxP validation alone is not sufficient. The fact that the software is used inside a clinical trial does not change its qualification — there is no investigational carve-out for SaMD.
Two patterns recur in vendor position statements that do not survive scrutiny. The first is the use of a "research use only" label either by the vendor or by the trial sponsor, which describes commercial intent but does not change the actual function of the software when deployed in a trial. The second is the "human-in-the-loop" defense, which describes an architectural feature — a qualified expert reviews the output before it is acted upon — but does not, on its own, displace qualification if the software is otherwise providing information used to take patient-level decisions. MDCG 2019-11 and current notified body practice are clear that disclaimers and review steps confirm a qualification narrative; they do not create one.
The CRO relationship deserves more explicit treatment than it usually gets. A vendor or CRO that builds, customizes, or operates the software is, in strict legal terms, the manufacturer if the software qualifies as a medical device. They carry the placing-on-the-market exposure under MDR Article 5 and the equivalent registration and listing exposure under FDA. But the regulatory consequences of misqualification land on the sponsor's MAA dossier, the sponsor's GCP inspection record, and the sponsor's development timeline. This asymmetry is why vendor- authored position statements that the sponsor adopts without independent scrutiny are an inspection liability — they shift the paperwork but not the underlying exposure. Sponsors that take qualification seriously own the position themselves, write it into the protocol, and treat the CRO's documentation as input rather than conclusion.
The honest answer about consequences is that most sponsors will not feel them immediately. The realistic failure mode is rarely a clinical hold, a recall, or any of the dramatic outcomes that get cited in conference talks. It is a quiet major finding during MAA review four or five years from now, when the trials currently being designed reach the dossier stage and reviewers start asking questions about the regulatory status of the software that generated key data. That delay is part of why the issue is so easy to underweight today. The team that took the qualification position is often not the team that has to defend it.
How hard the finding lands will depend on a few variables that are hard to predict. The first is the review pathway. A centralized EMA review with a reviewer whose agency has a strong digital health function will scrutinize software qualification in ways a national procedure for a less complex product may not. The second is the reviewer's familiarity with the software category in question. Imaging analysis pipelines and AI-driven decision-support are now well within the field of vision of major regulators; more specialized tools — radioligand dosimetry chains, niche biomarker analytics, novel digital endpoints — sit in regulatory territory where the questions are still being formed and where reviewer interpretation can still vary a lot. The third is enforcement appetite, which is itself a moving target. Authorities prioritize based on perceived patient risk, the precedential value of the case, and the resources available, and these calculations shift with each cycle of policy development.
The current period is a regulatory gray zone for software in trials, and gray zones do not stay gray indefinitely. MDCG has already revised 2019-11 once and is expected to issue further guidance on AI-enabled software, the EU AI Act's high-risk provisions are coming into force on a staggered timeline, the FDA continues to develop its AI/ML framework, and IMDRF working groups are active on adjacent questions. The direction of travel is toward more explicit expectations, not fewer. Sponsors and CROs whose current position depends on the absence of specific guidance are betting that the guidance does not arrive before their dossier does.
The practical consequence of all this is that the cost of taking a defensible qualification position is lowest now, when the protocol is being designed and the vendor is being selected, and increases at every subsequent stage. Remediation during the trial is expensive but feasible. Remediation during dossier preparation is expensive and disruptive. Remediation during MAA review is expensive, disruptive, and visible to the agency. Remediation after a major finding — repeating historical analyses through a qualified pipeline before pivotal data can be relied upon, or downgrading affected data from pivotal to supportive — can cost months to years on a registrational program, and on a competitive indication that is the operating consequence the steering committee will actually be discussing.
None of this means every borderline tool needs to be CE-marked. It means the qualification analysis is worth conducting deliberately, documented in a way the sponsor owns, before the trial starts. The investment is small. The downside protection is substantial, and it compounds across a program that uses the same vendor or the same software architecture in multiple trials.