Ana Pilar Olmos

Ana Pilar Olmos
One of the main challenges under Article 117 is determining when a device change becomes “significant.” Because the MDR and EMA provide no clear universal definition, manufacturers face a regulatory grey zone that can lead to unnecessary NBOp updates, delays, or compliance risks.
One of the more frustrating realities for manufacturers of integral drug-device combinations (iDDCs) is that neither the MDR nor the EMA framework offers a universal, codified definition of what constitutes a "significant" change to the device constituent.
Article 117 makes the consequence clear — a significant change triggers a new or updated Notified Body Opinion — but the threshold itself is left to interpretation, drawn from a patchwork of guidance documents, position papers, and industry practice. This grey area creates real exposure: manufacturers risk either over-reporting, and incurring unnecessary NBOp cycles with their associated cost and timeline impact, or under-reporting, and finding themselves out of compliance during inspection or variation review.
To close this gap, at Beyond Conception we apply a structured, risk-based framework that has already been put into practice by several manufacturers handling Article 117 submissions. The framework is grounded in the available guidance — in particular MDCG 2020-3, the Team-NB position paper, ISO 20069, and the EBE/EFPIA position paper on substantial design change — and translates their underlying logic into a workable decision workflow. Companies that have adopted it have used it both to avoid unnecessary NBOps and to defend non-significant determinations when challenged by regulators or notified bodies.
The logic itself is straightforward. When a change is assessed as significant, a new or updated Notified Body Opinion is required, and the variation timeline and dossier strategy need to be planned accordingly. When a change is assessed as non-significant, a clear justification supported by a risk assessment must be documented in Module 3.2.R of the eCTD, in line with EMA expectations. The latter is where most manufacturers underinvest. EMA's own Q&A makes clear that the absence of an NBOp does not relieve the applicant of the obligation to demonstrate, transparently and on the record, why the change does not warrant one. A weak or boilerplate justification in 3.2.R is, in practice, one of the most common findings in this area.
Applying a consistent framework gives manufacturers three concrete benefits: it avoids unnecessary NBOps where the change does not meet the significance threshold, it produces documentation that holds up under regulatory scrutiny, and it accelerates internal change-control decisions by giving cross-functional teams a shared reference point.
EMA 37991/2019 guidance requires a new/updated Notified Body Opinion for “addition or full replacement of the device or device part”.
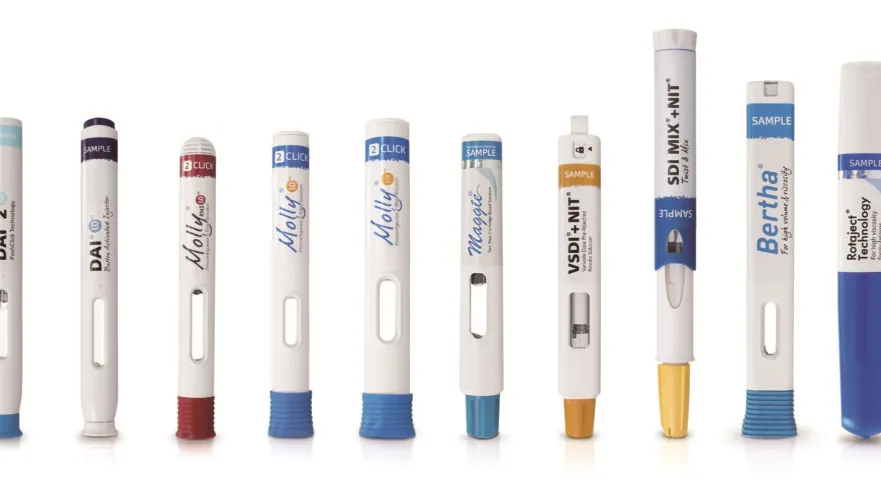
If a device or device part is being added to the product or fully replaced, then this will be require a new/updated NBOp, as per EMA guidance. The interpretation of when a device/device part is “fully” replaced is subject to interpretation, particularly as “device part” is not defined by the EMA.
Some examples that could meet such case would be:
And one example that is unlikely to meet such case can be:
Supporting Materials :